Crouzon syndrome is one of the most common craniosynostosis syndromes, leading to cranial and facial abnormalities. Typical phenotypic characteristics of Crouzon syndrome are cranial synostosis and brachycephaly, hypertelorism, low set ears and bulging eyes. The incidence of Crouzon syndrome is approximately 1 in 60,000 and diagnosis is usually made at birth, since prenatal diagnosis by ultrasound examination is not possible and standard radiography is usually inconclusive. As all other similar syndromes, it is inherited in an autosomal dominant mode, although the occurrence of de novo mutations in the developing embryo is quite common.
Crouzon syndrome
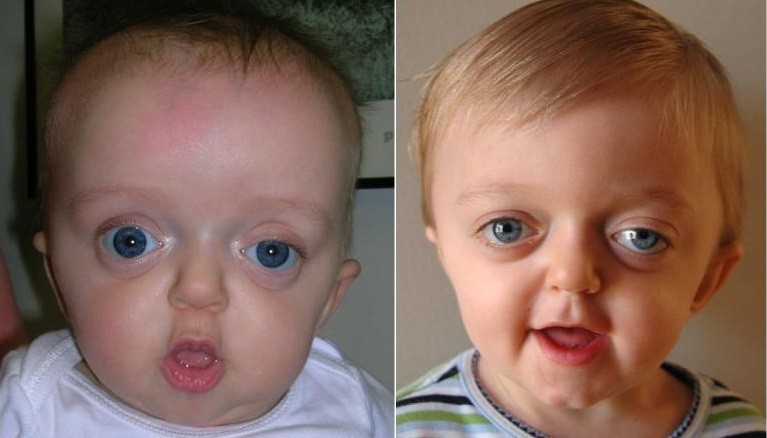
It is caused by specific mutations in the FGFR2 and FGFR3 genes and molecular genetic testing is frequently requested for confirmation of the diagnosis in an affected child. Advanced paternal age has been shown clinically to be associated with de novo mutations for Crouzon syndrome, Apert syndrome, Pfeiffer syndrome, Beare-Stevenson syndrome and Muenke syndrome. In any case, prenatal diagnosis for most craniosynostosis syndromes is not very common and is a sensitive issue, since the disorders are treatable through appropriate corrective surgery.
We perform automated bi-directional fluorescent DNA sequencing of exons 8 and 10 of the FGFR2 gene and of exon 7 of the FGFR3 gene, allowing for the detection of more than 90% of pathological mutations for the disorder. Please refer to Crouzon Syndrome with Acanthosis Nigricans for combined testing.