Apert syndrome belongs to the craniosynostosis group of syndromes, presenting with craniofacial malformations, syndactyly of fingers and toes and fusion of bone structures (with possible presence of soft tissue). Of all craniosynostosis syndromes, it is the one with the highest risk for mild to moderate mental retardation. Like all other syndromes in this group, it is an autosomal dominant disorder, although the majority of cases are sporadic. It is the result of mutations in the FGFR2 gene, the majority of which occur in exons 8 and 10 of the gene.
Apert syndrome
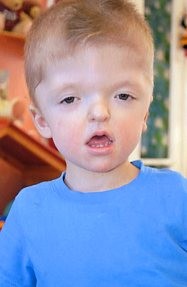
Advanced paternal age has been shown clinically to be associated with de novo mutations for Crouzon syndrome, Apert syndrome, Pfeiffer syndrome, Beare-Stevenson syndrome and Muenke syndrome. In any case, prenatal diagnosis for most craniosynostosis syndromes is not very common and is a sensitive issue, since the disorders are treatable through appropriate corrective surgery.
We perform automated bi-directional fluorescent DNA sequencing of exons 8 and 10 of the FGFR2 gene, allowing for the detection of more than 90% of pathological mutations for the disorder.