Η ομάδα των παθήσεων Charcot-Marie-Tooth αποτελεί συνολικά τα πιο κοινά κληρονομικά νευρολογικά νοσήματα και χωρίζεται σε πολλές υποκατηγορίες (π.χ. CMT1, CMT2, CMT4, CMTX, κ.α) και οφείλεται σε μεταλλάξεις πολλών γονιδίων, που ρυθμίζουν τη φυσιολογική λειτουργία των περιφερειακών νεύρων.
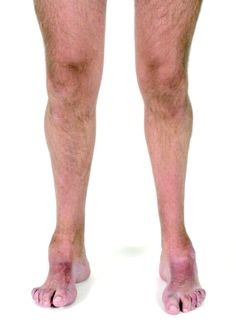
Ένα τυπικό χαρακτηριστικό της νόσου είναι η απώλεια του μυϊκού ιστού και της αίσθησης της αφής κυρίως στα κάτω άκρα αλλά εκδηλώνεται και στα άνω άκρα όταν βρίσκεται σε προχωρημένο στάδιο. Η νόσος διακρίνεται σε διαφορετικούς τύπους, που ο καθένας διαφέρει στον τρόπο κληρονομικότητας, στην ηλικία εκδήλωσης συμπτωμάτων και στη σοβαρότητα αυτών καθώς επίσης και στην εξέλιξη της νόσου. Η πιο συχνή και ‘κλασσική’ μορφή είναι ο τύπος CMT1A (συχνότητα 1-2/10.000 άτομα), που κληρονομείται με επικρατή τρόπο και οφείλεται σε βλάβες του γονιδίου PMP22 στη θέση 17p11.2. Η CMT1A οφείλεται σε >99% των περιπτώσεων σε διπλασιασμό του γονιδίου PMP22, βλάβη που είναι απόλυτα διαγνωστική για τη νόσο.