Prader-Wilil (PWS) syndrome is caused by the deletion or defects in the expression-methylation of one or more genes in the 15q12 paternal copy (the region is maternally inherited only, i. e. the paternally contributed region is absent). It is characterized by reduced fetal movement, muscular hypotonia, mental retardation, short stature, hypogonadism, obesity and small limbs. The majority of cases are sporadic, with an incidence frequency of 1/25.000 births.
Prader-Willi / Angelman syndromes
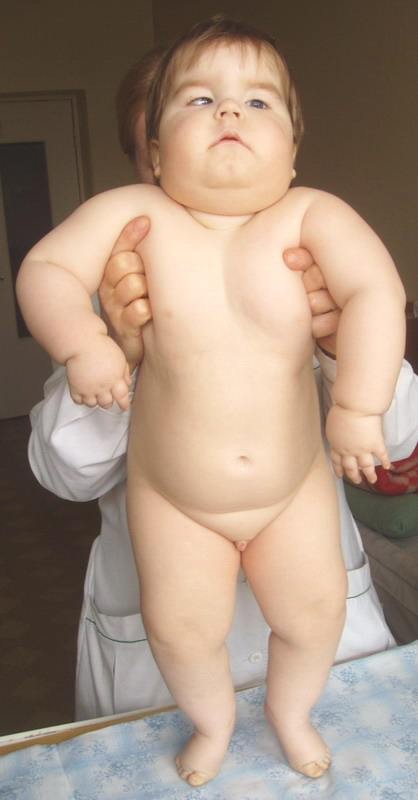
Angelman syndrome (AS) is similarly caused by the deletion or defects in the expression-methylation of one or more genes in the 15q12 maternal copy (the region is paternally inherited only, i. e. the maternally contributed region is absent). Mutations of the UBE3A gene are detected in ~10% of patients. The syndrome presents with severe developmental delay or mental retardation, speech impairment, gait ataxia or tremulousness of the limbs and a unique behavior (e.g. frequent laughing, smiling and excitability). Most clinical features of the syndrome may not become obvious until after the first 12-24 months and often the clinical diagnosis is made much later in childhood.

NOTE: Our laboratory participates with great success in the external quality assessment scheme organized by the European Molecular Genetics Quality Network (EMQN), which is periodically applied for Prader-Willi/Angelman syndrome testing.
The techniques applied (MLPA and MS-MLPA) enable us to detect genomic deletions, the methylation status of the 15p12 imprinted regions, as well as uniparental disomy (UPD), which together account for >99% of all PWS cases and ~80% of AS cases.
This technique is superior to FISH, due to its enhanced analytical sensitivity and reliability.