Ανάλυση όλων των γνωστών 48 γονιδίων, που συνδέονται με ένα ευρύ φάσμα κλινικών συμπτωμάτων καρδιομυοπάθειας
Οι καρδιομυοπάθειες, δηλαδή οι παθήσεις των καρδιακών μυών, αποτελούν μια σχετικά κοινή αιτία της καρδιακής ανεπάρκειας, που είναι συχνή αιτία θανάτου στις περισσότερες κοινωνίες. Υπάρχουν αρκετοί διαφορετικοί τύποι καρδιομυοπαθειών, πολλές από τις οποίες μπορεί να έχουν μια κληρονομική/γενετική αιτία.
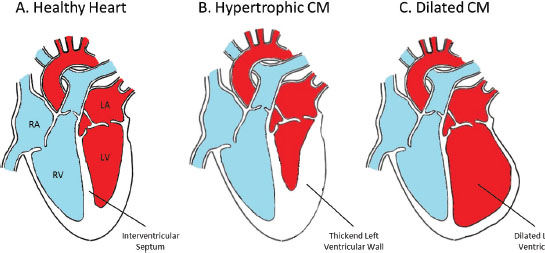
Οι δύο κύριοι τύποι είναι η διατατική καρδιομυοπάθεια (dilated cardiomyopathy – DCM) και η υπερτροφική καρδιομυοπάθεια (hypertrophic cardiomyopathy – HCM).
Πολλές διαφορετικές γενετικές αιτίες των μυοκαρδιοπαθειών έχουν ανακαλυφθεί και ο προσδιορισμός της υποκείμενης γενετικής αιτίας μπορεί να επηρεάσει τις αποφάσεις σχετικά με την ιατρική διαχείριση του πάσχοντα αλλά και τις επιλογές προληπτικού ελέγχου άλλων μελών της οικογένειας. Γενικά, πρέπει να δίνεται ιδιαίτερη προσοχή στο προσωπικό και οικογενειακό ιστορικό πριν συσταθεί η εφαρμογή της γενωμικής ανάλυσης για καρδιομυοπάθειες σε ένα μεμονωμένο ασθενή ή στην οικογένεια.
Ο καθορισμός της γενετικής αιτίας της καρδιομυοπάθειας μπορεί να είναι σύνθετη. Υπάρχουν πολλά διαφορετικά γονίδια και πολλές διαφορετικές μεταλλάξεις που εμπλέκονται στην κληρονομική προδιάθεση για καρδιομυοπάθεια.
Η αποκάλυψη της γενετικής αιτίας είναι περαιτέρω περίπλοκη επειδή υπάρχει σημαντική φαινοτυπική επικάλυψη μεταξύ των διαφόρων τύπων καρδιομυοπαθειών και επίσης σημαντική επικάλυψη στις γενετικές αιτίες μεταξύ των υποτύπων. Για παράδειγμα, τουλάχιστον 13 γονίδια έχουν ενοχοποιηθεί τόσο για HCM όσο και DCM και μπορεί ένα άτομο που φαινομενικά πάσχει από HCM να μη είναι δυνατό να διαγνωστεί με καρδιακή νόσο μέχρις ότου η ασθένεια είναι πολύ προχωρημένη και εκφραστεί τελικά ως DCM.
Επιπλέον, διάφοροι τύποι καρδιομυοπαθειών μπορεί να εκδηλώνονται μέσα σε οικογένειες, με μερικές οικογένειες να παρουσιάζουν πάσχοντες αποκλειστικά και μόνο από έναν τύπο καρδιομυοπάθειας, όπως HCM, ενώ σε μια άλλη οικογένεια η ίδια γενετική μεταβολή μπορεί να προκαλεί διαφορετικά είδη καρδιομυοπάθειας σε διάφορα μέλη της οικογένειας, όπως DCM ή HCM. Ακριβώς λόγω αυτής της επικάλυψης στα γενετικά αίτια, στα συμπτώματα και την εκδήλωση της ασθένειας για διαφορετικούς τύπους καρδιομυοπάθειας, ο κλινικός ιατρός μπορεί να συστήσει τον γενωμικό έλεγχο για όλα τα γνωστά τα γονίδια που σχετίζονται με τους διαφορετικούς υποτύπους καρδιομυοπαθειών σε ένα μόνο ολοκληρωμένο βήμα.
- Η υπερτροφική καρδιομυοπάθεια (HCM)
Εκτιμάται ότι εκδηλώνεται με συχνότητα ~1/500 και χαρακτηρίζεται από υπερτροφία της αριστερής κοιλίας (LVH), ανωμαλίες των μυοκυττάρων και ίνωση. Η ηλικία εμφάνισης κυμαίνεται από την παιδική ηλικία έως την πρώιμη ενήλικη ζωή. Μεταλλάξεις σε διάφορα γονίδια έχουν ενοχοποιηθεί με HCM και μεταλλάξεις στα γονίδια MYH7 και MYBPC3 αντιπροσωπεύουν ~60-80% των γενετικών αιτιών. Γενικά, τα γονίδια που εμπλέκονται στην HCM κωδικοποιούν κυρίως πρωτεΐνες του σαρκομεριδίου, ενώ έχουν παρατηρηθεί συνδρομικές και μη-συνδρομικές γενετικές μορφές HCM. Τέλος, περίπου 2-5% των περιστατικών φέρουν πολλαπλές μεταλλάξεις.
- Η διατατική καρδιομυοπάθεια (DCM)
Χαρακτηρίζεται από αριστερή κοιλιακή διεύρυνση (LVE) με κανονικό πάχος τοιχώματος και με συστολική δυσλειτουργία, με κλάσμα εξώθησης (EF) μικρότερο από 45-50%. Ο επιπολασμός της νόσου υπολογίζεται σε ~2.700, αν και αυτό το ποσοστό μπορεί να είναι υπο-εκτιμημένο. Η ισχαιμική καρδιακή νόσος είναι η πιο κοινή αιτία DCM, αντιπροσωπεύοντας περίπου το ήμισυ όλων των περιπτώσεων. Από τις υπόλοιπες μη-ισχαιμικές περιπτώσεις DCM, περίπου 20-35% των περιπτώσεων εμφανίζουν οικογενειακό ιστορικό. Πολλά γονίδια έχουν ενοχοποιηθεί τόσο στις οικογενείς όσο και σποραδικές μορφές μη-ισχαιμικής διατατικής καρδιομυοπάθειας. Μεταλλάξεις στα τρία γονίδια LMNA, MYH7, και TNNT2 έχουν ταυτοποιηθεί σε τουλάχιστον 15% των περιπτώσεων, με μεταλλάξεις σε άλλα πολλά γονίδια να αντιπροσωπεύουν σωρευτικά ένα άλλο 10-20% των γενετικών αιτιών και συμπεριλαμβάνουν γονίδια που κωδικοποιούν σαρκομεριδιακές πρωτεΐνες της πυρηνικής και κυτταρο-σκελετική μεμβράνης, κ.α.. Στην πλειοψηφία των περιπτώσεων, οι μεταλλάξεις των γονιδίων που σχετίζονται με DCM οδηγούν σε μη-συνδρομικές μορφές, αν και αρκετά γονίδια μπορεί επίσης να συνδέονται με συνδρομικές μορφές DCM.
Γενικά, η πλειοψηφία των καρδιομυοπαθειών εκδηλώνονται και κληρονομούνται με τον αυτοσωματικό επικρατή τρόπο, που σημαίνει ότι άτομα με μετάλλαξη σε ένα μόνο από τα δυο αντίγραφα κάποιου γονιδίου έχουν ιδιαίτερα αυξημένο κίνδυνο να νοσήσουν κι όλοι οι πρώτου βαθμού συγγενείς ενός ασθενούς έχουν 50% κίνδυνο να κληρονομήσουν την ασθένεια. Επίσης, είναι σημαντικό να αναφερθεί ότι διαφορετικές μεταλλάξεις στο ίδιο γονίδιο μπορεί να οδηγήσουν στην εκδήλωση διαφορετικού τύπου καρδιογενετικού νοσήματος.
Πρόσφατα, η τεχνολογία μαζικής αλληλούχισης επόμενης γενιάς (NextGenerationSequencing-NGS) έχει πλέον καταστεί μια ιδιαίτερα αποτελεσματική διαγνωστική στρατηγική, με την παράλληλη ανάλυση ενός μεγάλου αριθμού γονιδίων που εμπλέκονται στις καρδιομυοπάθειες.
Ο γενωμικός έλεγχος που έχει αναπτύξει και προσφέρει η InterGenetics περιλαμβάνει το μαζικό γενωμικό έλεγχο των 48 γονιδίων, που συνδέονται με ένα ευρύ φάσμα κλινικών συμπτωμάτων καρδιομυοπάθειας, ανεξάρτητα από τον τρόπο κληρονομικότητας.
Εκτελούμε ανάλυση της αλληλουχίας του DNA, με τη τεχνική Next Generation Sequencing (NGS) ) σε ειδικό αναλυτή Genome Analyzer – Ion Proton, όλων των εξονίων και των περιοχών συρραφής ιντρονίων-εξονίων (splice sites) των 48 γονιδίων, που μας επιτρέπει με τη βοήθεια σύνθετων και εξειδικευμένων εργαλείων πληροφορικής την ανίχνευση >98% των παθολογικών μεταλλάξεων των γονιδίων.
Όπου αυτό είναι εφικτό ή/και αναγκαίο, εκτελείται και επιπλέον ανάλυση MLPA για την ανίχνευση ελλείψεων/διπλασιασμών του γονιδίου (βλέπε την τελική αντίστοιχη έκθεση αποτελεσμάτων).
Η εξέταση είναι ιδιαίτερα ευαίσθητη και σύνθετη, επομένως είναι απαραίτητο τα αποτελέσματα να εκτιμηθούν από εξειδικευμένη ομάδα κλινικών και μοριακών γενετιστών, που εγγυάται τη μέγιστη δυνατή ασφάλειά σας.
H InterGenetics δεν είναι απλά ένα εργαστήριο Γενετικής. Είμαστε μια σύνθετη ομάδα από κλινικούς και εργαστηριακούς γενετιστές και γι’αυτό μπορούμε να σχεδιάσουμε μαζί σας ή/και με τον θεράποντα ιατρό σας, την ενδεδειγμένη εξέταση για το πρόβλημά σας. Τα αποτελέσματα των εξετάσεων μας, συνοδεύονται πάντα με λεπτομερή κλινική εκτίμηση των ευρημάτων.
Η σωστή κλινική γενετική εκτίμηση του περιστατικού και η γενετική συμβουλή, τόσο πριν όσο και μετά την εξέταση, είναι απαραίτητη προϋπόθεση, ώστε να καθορισθεί η σωστή στρατηγική του εργαστηριακού ελέγχου και να μπορέσουν να αποδοθούν σωστά οι έννοιες του πάσχοντος και του φυσιολογικού.
Υπό την επιστημονική επίβλεψη του Καθηγητή Ιατρικής Γενετικής Κ. Πάγκαλου, η InterGenetics αποτελεί ένα πρότυπο ιατρικό κέντρο διαγνωστικής Γενετικής διεθνών προδιαγραφών, με ιστορία 40 ετών στην Ελλάδα.
Η InterGenetics έχει πιστοποιηθεί ως Ion Torrent™ Certified Service Provider for Ion AmpliSeq sequencing στην πλατφόρμα Ion Proton.